Computer Aided Drug Discovery
Following problems are identified in this field:
Nearly infinite variability of chemical space of drug candidates
Large size of the libraries (millions of compounds)
Large size of protein-ligand complexes and the derived computational cost
Accurate prediction of protein-ligand binding free energy remaining elusive
Solution: New physics-based scoring methods capturing complex physics of protein-ligand interactions are necessary.
Large size of the ligand libraries makes challenging ligand structure validation. In the libraries encountering millions of compounds it is practically impossible to visually inspect each ligand on potential structure errors. Structural errors make impossible reliable atomic partial charge assignment. Effort has to be applied to minimize the ballast of broken structures. Reliable ligand structure validation may benefit from applying chemical intelligence into the process of structure inspection. We use the ability of LocalSCF program to check integrity of the input structure. The program can read and write ligands in MOL2 file format and write CM2 / CM3 partial atomic charges directly to MOL2 file.
10,962,930 drug-like compounds were downloaded from ZINC database (http://zinc.docking.org/). Ligand structure validation was performed by using LocalSCF program on 1-CPU desktop computer and it took 48 hours to complete. The performance is basically limited by the latency of input/output operations. Having a faster hard drive would immensely speed up the validation.
The ligands for the validation test are provided in a single multiple-structure MOL2 file (ligands.mol2). The control file for LocalSCF (ligands.setup) needs only two keywords "0SCF NORESI". Keyword 0SCF tells the program to perform only the validation without starting QM calculation. Keyword NORESI tells the program to skip identification of amino acid residues. To start the computation type in:
LocalSCF.x ligands.mol2
When calculation is finished two files ligands-OK.mol2 and ligands-bad.mol2 are created. First MOL2 file contains the ligands which passed the validation test. This is a perfectly clean database for docking purposes. The second MOL2 file contains ligands which failed the test. It can be discarded.
Simulation identified 20,233 broken structures which represent 0.2% of the total number of the validated compounds. This translates to occurrance of 1 problematic structure per 5,000 correct structures. The low percentage of the errors speaks about high quality of the ZINC database. Though, even such clean database can benefit from additional easy-to perform clean up. The information about broken structures can be used to further refine the ligand structure generation tools.
The 20,233 broken ligands can be further categorized:
1,308 compounds with wrong assigned total charge
5,489 compounds have short interatomic contacts
3,885 compounds have jammed structure
8,100 compounds have stretched bonds
1,451 compounds have incorrect hydrogen atom assignment
Selected examples of problematic structures
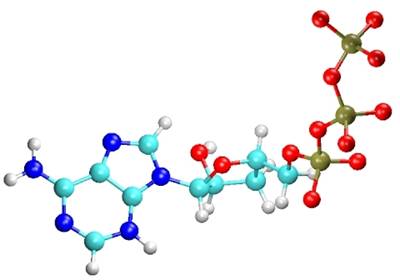
#11944231
Q(mol2) = -1
Q(LocalSCF) = -3
#05940603
an open-shell system (a radical)
#04648965
stretched bond
In reality it may be too difficult to fix every ligand structure in the database. With help of the LocalSCF program we can filter out the problematic structures removing them from the database so the resulting set will be 100% error-free.
Having prepared the ligand database next step would be to dock the ligands to the receptor. One can use their favorite docking software to accomplish this task.
Next comes the challenging step to score the ligands based on their binding free energy. To accomplish this step we use all-atom quantum mechanical calculations by means of the LocalSCF program.
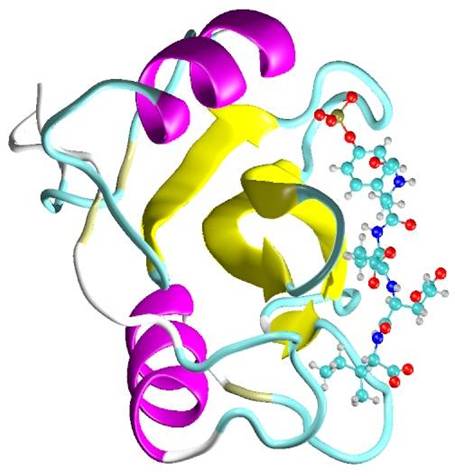
QM Docking of p56lck SH2 domain
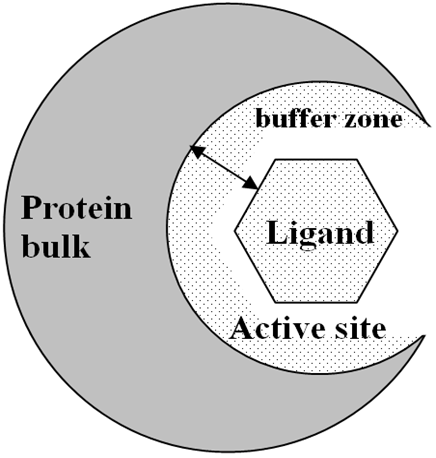
QM docking in LocalSCF program is based on a 2-layer approach in which ligand and protein active site are treated variationally while the distant part of the protein is represented by a frozen electron density matrix. Both layers in this model are treated at QM level. The electron density matrix corresponding to protein bulk is computed once and then reused with each ligand. The active site is computed self-consistently for every new ligand. Due to 2-layer approach the calculation time is reduced roughly by factor of 10. This approach is schematically represented in the following figures
See V. M. Anisimov, V. L. Bugaenko; J. Comp. Chem., 2009, 30 (5) 784-798 for further details.
20,000 drug-like compounds were docked to p56lck SH2 domain by using a traditional docking approach. Top 10 poses were saved for each ligand.
The resulting 200,000 protein-ligand complexes (1700 atoms each) were subjected to AM1 calculation at rigid geometry. This calculation took 32 days on 1 CPU. Correspondingly the simulation would be finished in 1 day on a 32-CPU cluster.
To obtain the effect of QM docking the resulting 10,000 top-scoring poses were subjected to ligand geometry optimization which took 17 days on 1 CPU.
The short-listed best binders can be subjected to rigorous QM free energy calculations using MM-QMSA approach. The most sophisticated free energy calculations can be performed based on QM MD.
Copyright (c) 2010 Victor Anisimov