|
|
| LocalSCF | Understanding the mechanisms of macromolecular systems builds on appreciation of rich physics of the underlying atomistic processes. It is well understood, that atomistic processes are governed by quantum-mechanical principles. | 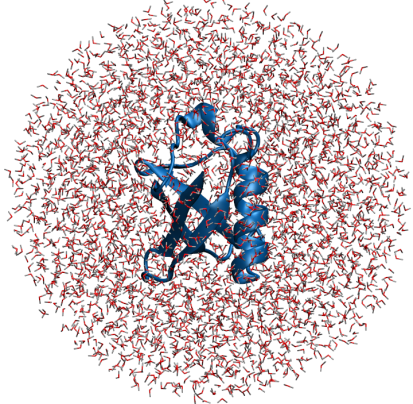 | To keep the low computational overhead it is necessary to use certain approximations to the QM scheme. LocalSCF is a linear scaling method utilizing local electronic properties of molecular systems. Due to its modest computational requirement LocalSCF opens the possibility to study thermodynamic properties of liquids and solids, drug-receptor interactions, enzyme catalysis, free energies, and provides insights into many processes which require rigorous sampling of potential energy surfaces by employing molecular dynamics. Ubiquitin in water; 12199 atoms; 20 ps QM MD ChemPhysChem, 2009, 10 (18) 3194 | | Unlike mechanical engineering with its foremost example of entirely computer-based design of aircraft Boeing-777, simulation in Chemistry and Biology will unlikely be able to substitute experiments in near future. In the face of Chemistry and Biology, Nature found a more efficient device to produce new materials. Nevertheless, simulation in Chemistry and Biology is immensely important because it provides atomic-level details not available from experimental studies. Simulations help understanding experimental data, and through that assist in designing new experiments and materials. read more | | | | Validation of 2-layer all-atom QM Method for Protein-Ligand Docking Difference in Total Energy as a function of buffer-zone thickness |  | 2-layer QM / QM method converges to the conventional 1-layer QM on buffer-zone thickness from 6 to 12 Å. Buffer zone is part of the protein located in close proximity to the bound ligand. Buffer zone and ligand are treated at the regular SCF level whereas the protein bulk carries frozen density matrix. The character of energy convergence suggests that electronic polarization can only be fully captured when the entire system is treated at QM level. J. Comp. Chem., 2009, 30 (5) 784. | | | | | Copyright (c) 2011-2012 Victor Anisimov | |
| |
|
